Impulse conduction in a single axon is fascinating to behold but, taken by itself, functionally limited. The full potential of the impulse is appreciated only by the functional changes it produces in a postsynaptic cell. Here we will examine the events which occur at these functional contacts known as synapses.
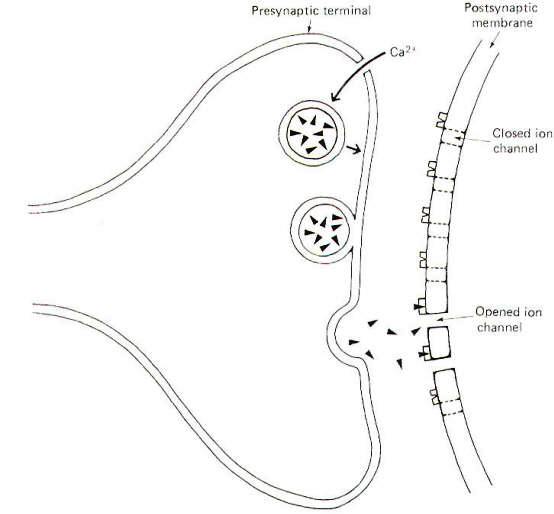
Neurons make functional contact with other neurons as well as with the cells of skeletal muscle, cardiac muscle, smooth muscle, and glands. The contacts neurons make with these cells are called synapses, a term meaning "connection" coined by the English physiologist Sherrington. The "connection" is actually an extracellular fluid-filled synaptic cleft separating the nerve cell membrane from the postsynaptic cell membrane (Fig-1). This narrow cleft is typically 20 nm wide, a span sufficiently great to bring to an abrupt halt the transmission of impulses.
 | |
| Fig-1 |
The signal must bridge this cleft in order to influence the postsynaptic cell. This is effectively produced at chemical synapses by the release of chemical neurotransmitters from the presynaptic terminal, which diffuse within microseconds across the cleft to specific receptor sites on the postsynaptic cell membrane. The neurotransmitter-receptor site interaction then causes specific ion channels to open on the postsynaptic membrane, triggering ionic fluxes which either depolarize or hyperpolarize the membrane. Excitatory synapses depolarize postsynaptic membranes while inhibitory synapses hyperpolarize them.
Depolarization of muscle cell membranes leads to contraction while depolarization of a postsynaptic neuron leads to the propagation of impulses on its axon. Conversely the hyperpolarization of a muscle cell membrane prevents contraction, while hyperpolarization of postsynaptic neurons prevents impulse conduction.
In order to allow presynaptic terminals to effectively control postsynaptic cells, it is necessary to quickly inactivate the released neurotransmitters after they have activated receptor sites, otherwise the postsynaptic cells will continue to be stimulated or inhibited longer than desired. Only by having the postsynaptic response occur immediately following firing of the presynaptic terminal, and not for prolonged periods afterwards, can the presynaptic neuron maintain this control. Thus the postsynaptic cell can be driven to continual action by repetitive firing of the presynaptic neuron or brought to an abrupt halt by the termination of presynaptic input.
Released neurotransmitters are rendered inactive by any or all of three means. At some synapses the transmitters are rapidly and actively reabsorbed by the presynaptic neuron for possible release a second time, a process called reuptake. A second means of inactivation is by the enzymatic degradation of the neurotransmitters by hydrolyzing enzymes which are present in the synaptic cleft or on the postsynaptic membrane. Still a third means of inactivation is for the transmitters to diffuse out of the synaptic cleft and away from the receptor sites.
While the term synapse is often used to describe all functionally active neuron contact with receptor cells, certain additional terms are in common use. For example, the neuron-neuron contact is called a neuronal synapse. while the neuron-skeletal muscle cell contact is called a neuromuscular or myoneural junction. The contacts made by nerve cells with cardiac muscle, smooth muscle, and gland cells are all neuroeffector junctions.
The particular physiological response produced in a receptor cell is determined by (1) the type of neurotransmitter released, (2) the quantity released, (3) the type of receptor site encountered, and (4) the particular function of the receptor cell. The neurotransmitter acetylcholine (ACh), for example, causes an increase in the contractility of the smooth muscle of the stomach, while norepinephrine (NE) produces decreased activity in this same muscle. Sufficient ACh is typically released from a single discharge at the neuromuscular junction to produce contraction of the skeletal muscle cell. In certain disease conditions, however, insufficient ACh is released to reach the excitation threshold of the cell and it does not contract. Furthermore, receptor cells may contain different types of receptor sites for the same neurotransmitter. For example, NE binds with alpha receptor sites on some vascular smooth muscle cells to produce vasoconstriction while binding with beta receptor sites on others to produce vasodilation. Ultimately, of course, the response capability of a receptor cell is determined by its function in the body. Muscle cells can either contract or relax, glandular cells can either secrete or not, and nerve cells can be made either to conduct impulses or not.
Certain synapses in invertebrates are electrically mediated rather than dependent on chemical transmission. The clefts at such synapses are usually narrower than those at chemical synapses. The electrical currents associated with the impulse at the presynaptic terminal spread across the cleft to directly stimulate the postsynaptic membrane electrically. However, in the overwhelming majority of mammalian and other vertebrate synapses the cleft is too wide for electrical transmission, and chemical transmission is required to bridge the gap.
There is strong evidence to implicate certain chemicals as neurotransmitters at synapses. Others, known as putative transmitters, are also suspected to act in this way, but the evidence supporting their participation is not as complete. It is generally agreed that for a substance to qualify as a neurotransmitter it must satisfy the following criteria:
1 The substance and the enzymes necessary for its synthesis are present in the neuron.
2 Impulses reaching the presynaptic terminals will release the substance.
3 Systems exist for the rapid inactivation of the substance.
4 Local application of the substance produces changes similar to those produced by synaptic release.
5 Drug-induced responses to both locally applied and synaptically released substances are similar.
Acetylcholine, norepinephrine, and dopamine (DA) are chemicals which have fulfilled all of these criteria. Nevertheless, several additional physiological chemicals have met some but not all of the criteria and are also suspected to function as neurotransmitters. ACh, NE, and DA have been identified in both the peripheral and central nervous systems while the others listed in Table-1 are thought to operate in the CNS only.
It is presently not known whether the enzymes necessary for the synthesis of the various neurotransmitters are themselves synthesized at neuronal endings since no ribosomes have been detected in axons even with the aid of the electron microscope. Nevertheless, the enzymes are found there. There is some evidence to suggest that they are synthesized in the soma and sent by axonal transport to the neuronal endings. However, it might also be that they are synthesized at some point along the axon by mechanisms as yet unknown. In any event neurotransmitters are most certainly synthesized in the neuronal endings since the rate of axonal transport is much too slow to account for the rapid replenishment which is necessary to prevent synaptic fatigue (neurotransmitter depletion) even in a slowly firing neuron.
| Table-1 Known and Suspected Neurotransmitters | |
| Acetylcholine (ACh) | |
| Norepinephrine (NE) | |
| Dopamine (DA) | |
| Prostaglandins | |
| Serotonin | |
| Histamine | |
| Glycine | |
| Aspartic acid | |
| Glutamic acid | |
| y-Aminobutyric acid (GABA) | |
Much of our knowledge about synapses is based on observations of the spinal motor neuron. The neuronal synapse is composed of a presynaptic terminal (PST), a synaptic cleft, and a postsynaptic membrane. These synapses are often classified according to where they contact the receptor neuron. Accordingly we have axodendritic, axosomatic, and axoaxonic synapses depending on whether the PSTs contact a dendrite, soma, or axon. Axoaxonic synapses are rare, with dendritic and somatic contacts being the general rule. Often hundreds to thousands of axodendritic and axosomatic synapses will occur on a single motor neuron.
The arrival of an impulse at the presynaptic terminal causes the release of transmitter and its subsequent diffusion across the cleft where it activates postsynaptic receptor sites opening specific ion channels (Fig-1). At an excitatory synapse, ionic fluxes through these channels tend to depolarize the membrane, while different patterns of ionic flux hyperpolarize the membrane at inhibitory synapses.
Interaction with receptor sites at excitatory synapses opens Na+ and K+ channels, thereby increasing the permeability of the postsynaptic membrane to each of these ions. Consequently Na+ tends to diffuse into the cell while K+diffuses outward, each following its own chemical gradient. However, the inward Na+ current is greater than the outward K+ current, causing the postsynaptic membrane to depolarize. Thus the postsynaptic membrane potential is no longer resting and is now called an excitatory postsynaptic potential (EPSP) (Fig-2). The potential is called excitatory because the membrane potential is closer to the excitation threshold than it was in the resting state.
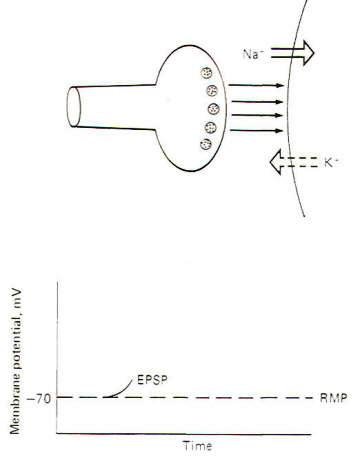 |
Fig-2
|
If the EPSP is produced by a single, not a repetitive, volley of transmissions across the synapse, a small EPSP called a local response will be produced which will decay over a period of 15 ms or so to the resting state as the Na+ and K+ channels resume their normal permeabilities and neurotransmitter is inactivated. Recall that this normal permeability has K+ diffusing outward more readily than Na+ diffuses inward, thus repolarizing the membrane to the resting state.
An inhibitory synapse produces effects just opposite to those at the excitatory synapse. Here the action of the transmitters on the receptor sites is to open those ionic channels which hyperpolarize the postsynaptic membrane. Typically these are the K+ and Cl- channels. Recall that the chemical gradients of these two ions are such that K+ diffuses outward while Cl- diffuses in. This combination of ionic fluxes hyperpolarizes the membrane so that the internal potential becomes even more negative than the resting state. Consequently an inhibitory postsynaptic potential (IPSP) is established (Fig-3). The potential is caned inhibitory because the membrane potential is even farther from the excitation threshold than in the resting state.
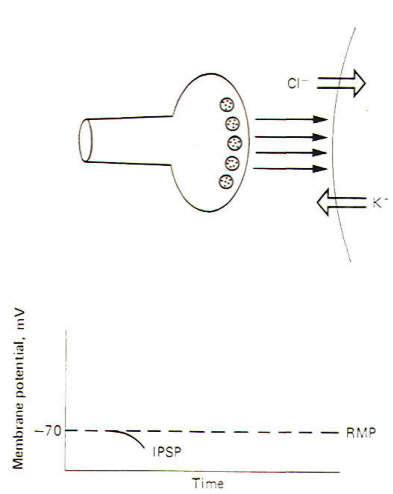 |
Fig-3
|
Before an action potential can develop in the receptor cell, the membrane must depolarize to the excitation threshold. In the spinal cord alpha motor neuron. this threshold is about -40 mV in the dendrites and soma but approximately -59 mV in the initial segment of the axon, the axon hillock. Since the resting membrane potential in all three regions is the same (approximately -70 mV), the axon hillock is easily the most excitable part of the neuron as it need depolarize only 11 mV in order to reach excitation to produce an action potential. Thus this is the point of impulse generation in the motor neuron.
Since the majority of synapses on the motor neuron are axodendritic and axosomatic, one must ask the question, "How does depolarization at a distant excitatory synapse cause depolarization of the membrane in the axon hillock?" The answer lies in the spread of a depolarizing electrotonic current from each synapse as it depolarizes. An examination of the activity at a single synapse will serve to introduce the point. When the receptors at an excitatory synapse are activated by neurotransmitter and ion channels open which favor a net influx of positive charges, the postsynaptic membrane depolarizes slightly. It has been estimated that a single synapse firing once on the motor neuron releases enough neurotransmitter to establish an EPSP of approximately 100 to 200 µV. As this is obviously much too weak to reach excitation, no action potential is generated. Further, as we pointed earlier. this miniature EPSP will decay back to the resting membrane potential level within 15 ms if no additional firings occur at the synapse. Nevertheless, during the EPSP the interior of the postsynaptic membrane is temporarily less negative than the neuroplasm at a distance from the synapse. Accordingly, a passive electronic (local) current spreads from the less negative to the more negative region and out through the adjacent membrane as a depolarizing capacitive current. The length constant of this current is usually sufficient to reach from even the most distant dendrite to the soma and axon hillock. This means that while the strength of this outwarddirected capacitive current decreases away from the synapse. there is still some left to help depolarize the axon hillock.
Now while the EPSP produced by a single synapse firing once is insufficient to produce a strong enough electrotonic current to depolarize the axon hillock to the excitation threshold, many separate synapses firing simultaneously, or even a single one firing repetitively at a very high rate, are sufficient to do so. The former pattern is spatial summation and the latter is temporal summation. Thus the membrane potential on the axon hillock can be depolarized to the excitation threshold and subsequently give rise to an action potential by either spatial or temporal summation of the synaptic EPSPs.
Spatial summation is the establishment of a summated EPSP by the simultaneous firing of many synapses distributed over the dendrites and soma. If enough of them fire at the same time, the local EPSPs will summate to produce an electrotonic current of sufficient strength to depolarize the axon hillock to the excitation threshold. In this way the synaptic potentials at distant dendritic sites contribute to the production of an action potential on the hillock through the instantaneous spread of the electrotonic current (Fig-4). If an insufficient number of synapses fire simultaneously, the summated EPSPs will not reach the excitation threshold and a local response, but no action potential will be seen. This local response is graded while the action potential is not. This means that the amplitude of the summated EPSPs varies directly with the number of synapses simultaneously firing. Thus, below the excitation threshold, increasing the number of presynaptic terminals firing will increase the amplitude of the potential while a decrease in the number firing will decrease it. On the other hand. if the number of PSTs firing is sufficient to reach the excitation threshold. a nongraded action potential will be generated. "Nongraded" means that the amplitude of the action potential will be the same any time enough synapses fire to reach threshold. Even if twice this number fire the amplitude will not change. Thus the action potential is an all-or-none response.
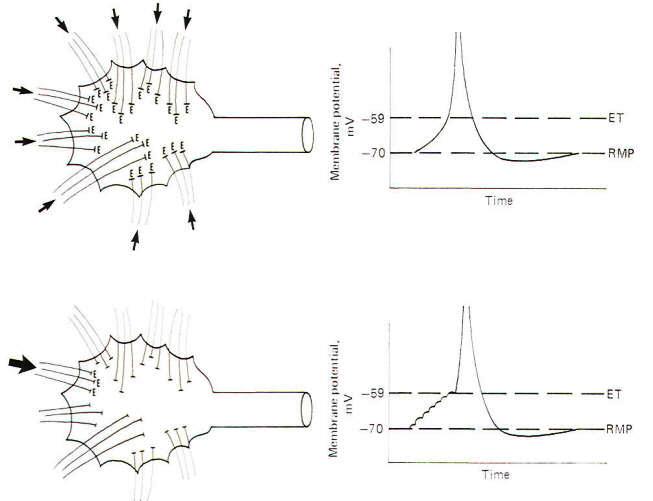 |
Fig-4
|
Once an action potential is generated on the hillock, it self-propagates down the length of the axon. That is, a local electrotonic current, initiated during the reversed stage of the action potential, travels through the axoplasm and out through the adjacent membrane as a depolarizing capacitive current bringing the adjacent membrane to the excitation threshold, establishing a second action potential, and so on. A little reflection will make it clear that the action potential, when it does occur, will start on the axon hillock rather than on the soma or dendrites. Even though the summated electrotonic current generated by simultaneously firing synapses depolarizes dendritic and somatic membranes on its way to the hillock. only 11 mV of depolarization are required for excitation here while approximately 30 mV are necessary on the less excitable dendrites and soma. Thus it is the first region to produce an action potential.
One should also be aware that the graded local response lasts longer than the increase in Na+ conductance gNa+ which caused it, since restoration by K+ outflow takes a little time. This is a significant feature because it allows the postsynaptic neuron an alternative to spatial summation by which it can produce an action potential and a propagated impulse. This alternative method is temporal summation.
Temporal summation is the establishment of a summated EPSP by the repetitive firing at a high rate of a single excitatory synapse. Recall that the approximate 1 ms required for a single action potential imposes an upper limit of about a thousand impulses per second on a neuron's firing rate. Now since the EPSP from a single synaptic firing lasts up to 15 ms. it is apparent that if a single synapse fired repetitively at a high rate. the EPSPs would summate, producing a greater degree of depolarization than would be caused by a single firing. Thus the potentials are summed over time and the process is called temporal summation. If the depolarization produced by the temporal summation of the synaptic EPSPs is sufficient to reach excitation. an action potential is produced in the axon hillock. Because a single EPSP may start to decay before the next one summates, the rise to the excitation threshold can be pictured as a steplike progression (Fig-4).
A single motor neuron might receive presynaptic innervation from many hundreds or thousands of input neurons. Some of these synapses will be excitatory, while others will be inhibitory. We have previously described how excitatory synapses give rise to EPSPs while IPSPs are produced at inhibitory synapses. It should be apparent that the amplitude of the summated EPSP on the axon hillock will be decreased by the hyperpolarizing effect of several simultaneously firing synapses. Clearly then the state of the membrane potential on the soma and axon hillock of the motor neuron at any given time is determined by the number, type, and firing frequency of its incoming synapses. Only when this "integrated" potential exceeds the excitation threshold of the hillock will an action potential occur.
Thus several combinations of events exist that can produce an action potential in the motor neuron. These are (1) a single, or at least a very few, excitatory synapses firing simultaneously at a high rate with no inhibitory synapses firing simultaneously. (2) many excitatory synapses firing simultaneously at multiple locations on the neuron with no simultaneously firing inhibitory synapses. and (3) increasing the amplitude of the temporally or spatially summated EPSP in order to overcome the hyperpolarizing effect of inhibitory synapses firing simultaneously. In this latter case, if the inhibitory synapses considered by themselves could have produced an IPSP 3 mV more negative than resting (i.e., -73 mV), the summated EPSP produced by the excitatory synapses would need to be sufficiently increased to depolarize the hillock membrane by 14 mV rather than 11 mV in order to reach threshold and generate an action potential. A quantity of 3 mV is required to overcome the IPSP and another 11 mV to reach -59 mV, the excitation threshold.
It must be recognized that the membrane potential on the axon hillock is not a simple algebraic summation of the number of excitatory and inhibitory synapses firing at any given time. The relative position of the synapses on the dendritic tree of the motor neuron and the timing of their firing can have profound effects on this "integrated" potential. For example, if a single inhibitory synapse is located near the point where a dendrite joins the soma while a single excitatory synapse is located near the periphery of this same dendrite, and both are fired simultaneously, the IPSP will have a potent effect toward decreasing the EPSP, resulting in a seriously decreased EPSP on the axon hillock. If, however, the position of the two synapses is reversed with the excitatory synapse between the inhibitory synapse and the soma, the simultaneous firing of both does not cause much reduction in the hillock EPSP. Similarly, slight variations in the relative timing of the firings of the synapses can have significant effects on their ability to influence the hillock potential.
If all the synapses converging on a single motor neuron were to fire just once with the overwhelming majority being excitatory, the neuron would depolarize to the excitation threshold of the axon hillock and a single impulse would be generated which would travel out along the axon to its terminals. However. if no synapses, either excitatory or inhibitory, were to fire, the membrane potential of the postsynaptic neuron would be considered truly resting (Fig-5). It is possible, however, for several synapses to fire repetitively at a low enough rate and with sufficient timing to maintain a summated EPSP on the axon hillock several millivolts closer to the excitation threshold than the resting state. If this summated EPSP is 5 mV above the RMP, the neuron is said to have a central excitatory state (CES) of 5 mV. Recognize that a neuron which is maintaining a central excitatory state of 5 mV is in a more excitable condition than if it were in the resting state since it need depolarize only 6 mV more to reach threshold (Fig-5).
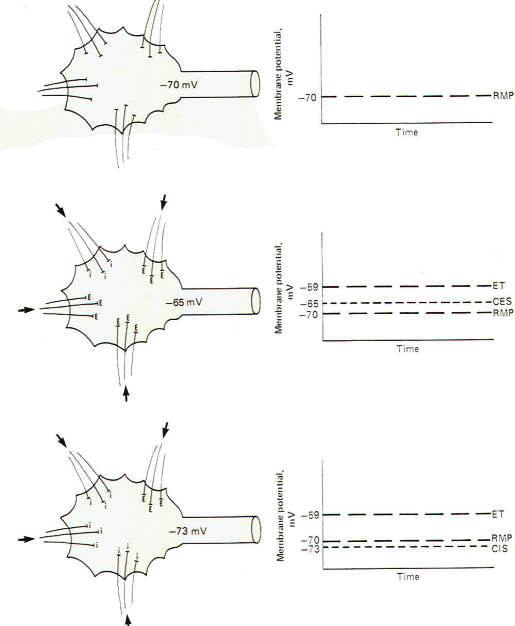 |
Fig-5
|
It is also possible for a neuron to maintain a central inhibitory state (CIS) by the appropriately timed repetitive firing of inhibitory synapses. Of course in this case a greater degree of subsequent excitatory input would be required to reach the excitation threshold (Fig-5).
Thus. by a steady subthreshold repetitive excitatory input, neurons can be maintained in a "ready" condition so that they can quickly respond to additional input and fire rapidly. One can visualize the importance of this capability, for example, in the activation of escape mechanisms in animal muscle systems. Similarly, the excitability of a neuron can be decreased by the maintenance of a CIS.
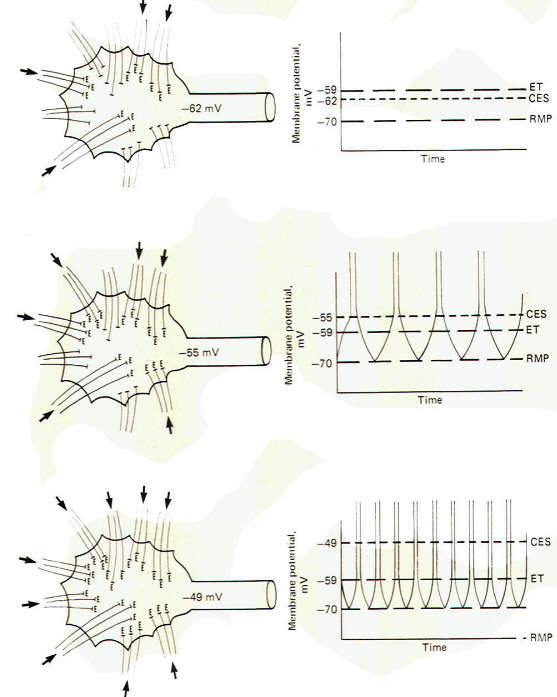 |
Fig-6
|
The upper drawing in Fig-6 shows a motor neuron with sufficient low level repetitive excitatory input to maintain a central excitatory state of 8 mV. That is. the membrane potential is being held at -62 mV. which is 8 mV above the resting state. Of course, the motor neuron does not generate an action potential and subsequent impulse, as the excitation threshold is not reached. It is merely in a more excitable state. The middle and lower drawings in Fig-6 show the effect of maintaining a central excitatory state above the excitation threshold. In the middle drawing. sufficient excitatory input has raised the CES to 15 mV, 4 mV above the excitation threshold. Accordingly, an action potential will be generated on the hillock which propagates down the axon as an impulse. Now let's look more closely at this action potential. Following the depolarization phase in which the reversed potential is established, the membrane needs to return to the polarized resting state in order to be excitable once again. This repolarization is of course caused by a strong outward potassium current IK+ which drives the membrane toward the potassium equilibrium potential EK+. However, following repolarization, the hillock membrane once again begins to depolarize because of the steady ongoing suprathreshold repetitive excitatory input which drives the CES back to 15 mV. Of course as soon as the threshold point is passed (at -59 mV), sodium conductance gNa+ dramatically increases, producing a strong inward sodium current INa+ and giving rise to a second action potential. The neuron will continue to fire impulses at a steady rate as long as the same level of CES is maintained by the excitatory synaptic input.
Increasing the level of this input, and hence increasing the CES, will produce an increase in the neuron's firing rate. In the bottom drawing in Fig-6 the CES is increased to 21 mV, maintaining the hillock membrane potential at -49 mV. Notice that the number of excitatory synapses firing onto the postsynaptic neuron has increased over that shown in the middle drawing, bringing about the increase in the CES. Notice also that the firing rate of the second neuron has correspondingly increased because of the increased CES. This increased firing rate is a direct result of an increase in the speed of depolarization following each action potential. Thus the firing rate of a continually stimulated neuron is a function of the degree to which this stimulation maintains a CES in excess of the neuron's excitation threshold.
A number of factors have been identified which influence transmission at the synapse. These will be discussed here.
The Bell-Magendie Law and One-Way Conduction
Whenever an action potential is generated in the axon hillock, an impulse is generated which travels out over the axon toward its terminal endings. This is described as an orthodromic (running forward) impulse. At the same time, an antidromic (running backward) impulse is generated which spreads back over the soma, and to a certain extent, out to the dendrites. The orthodromic impulse carries the potential for modulating activity in the postsynaptic neuron through synaptic transmission. That is, it causes transmitter release and subsequent excitation or inhibition of the postsynaptic membrane. On the other hand, the antidromic impulse has no such potential for modulation. There is no way for the postsynaptic membrane to communicate with the presynaptic terminal through "backward" transmission at the synapse. There are no vesicles releasing transmitter at the postsynaptic membrane. Thus transmission occurs in only one direction at the synapse, from the presynaptic terminal toward the postsynaptic membrane. This is the Bell-Magendie law.
Synaptic Delay
The smallest nonmyelinated type C nerve fibers conduct impulses as slow as 0.2 m/s. whereas large myelinated type A fibers conduct impulses at velocities up to 120 m/s. However, regardless of the conduction velocity up to the synapse, the speed at which the postsynaptic cell is stimulated is limited by the time required for the events at the synapse, called synaptic delay. Because of their ease of access, the only synapses which have been extensively studied with regard to synaptic delay are those incorporated in spinal reflexes. The average synaptic delay time at these synapses is approximately 0.5 ms. This delay represents the time it takes to release and diffuse neurotransmitter across the synaptic cleft and for the receptor sites to become activated. The reader should be aware that the 0.5 ms time does not necessarily hold for all synapses, as we have no valid data for synaptic delay times in the brain, for example. However, it probably serves as a good first approximation.
Synaptic Fatigue
If a presynaptic terminal fires and releases neurotransmitters faster than it can synthesize and store new transmitter, the synapse will soon be depleted of stored transmitter and stop functioning. This stoppage is called synaptic fatigue. It has been estimated that fatigue would occur within a few seconds if resynthesis were suddenly stopped and the synapses were fired at a high rate. There may be enough transmitter available for up to 10,000 transmissions by a single PST before it would become totally fatigued under these conditions. Nevertheless, recognize that under normal circumstances, synapses can fire as many as 1000 times per second for long periods of time and maintain a reuptake and resynthesis rate sufficient to prevent fatigue.
Ca2+ and Mg2+ Concentrations and Synaptic Transmission
Up to now we have implied that the arrival of an impulse at the presynaptic terminal is sufficient to release neurotransmitter from presynaptic vesicles in all circumstances. In fact, the amount of neurotransmitter released by the arrival of the impulse at the PST depends on the concentrations of Ca2+ and Mg2+ in the solution bathing the terminal. If the Ca2+ concentration is reduced or the Mg2+ concentration is increased, the amplitude of the synaptic potential is progressively reduced.
The release of neurotransmitter is dependent upon the entry of Ca2+ to the presynaptic terminal. When the normal sodium and potassium currents are blocked with tetradotoxin (TTX) and tetraethylammonium (TEA), respectively, there is a measurable inward current remaining in stimulated axon terminals that has been shown to be totally dependent on the concentration of external Ca2+. These data indicate that Ca2+ enters the presynaptic terminals on the arrival of an impulse.
The inhibitory effect of Mg2+ on transmitter release appears to be due to its antagonistic effect on Ca2+ entry. It apparently competes with Ca2+ for membrane sites and thus interferes with the normal inward Ca2+ current. Thus it seems that the arrival of the impulse at the PST causes transmitter release indirectly by first moving Ca2+ into the terminal. Then, by some still unknown mechanism, this causes transmitter release. Evidence also suggests that the greatest part of the synaptic delay is taken up by the time required for Ca2+ entry and transmitter release.
In experiments that exploited the fact that transmitter release can be greatly reduced by manipulating the Ca2+ and Mg2+ concentrations, some significant features of the mechanisms of transmitter release were discovered. For example. at very low levels of transmitter release, the amplitude of the synaptic potential varies on repeated observations as a multiple of some irreducible unit size. It has been postulated that this unit amplitude results from the release of a "quantum" of neurotransmitter. It is likely that this quantum relates to the number of neurotransmitter molecules in a single synaptic vesicle. The "miniature potentials" produced by the release of a quantum of transmitter may be the building blocks upon which the normal synaptic potential is built when multiple vesicles release transmitter upon the arrival of an impulse at the PST when the concentrations of Ca2+ and Mg2+ are normal.
pH and Synaptic Transmission
Synaptic transmission is highly pH-dependent. Increasing the pH increases transmission while decreasing the pH decreases it. This is particularly apparent in brain synapses where alkalosis of 7.8 (normal. 7.4) increases excitability of neural pathways to the point of bringing on cerebral convulsions, while a decrease in pH to less than 7.0 decreases excitability to the point of coma. The latter is always seen in severe uremic or diabetic acidosis.
Drugs and Synaptic Transmission
A number of drugs are available which can alter transmission at the neuronal synapse. A few examples are caffeine (found in coffee) and theophylline (found in tea). which are known to increase synaptic excitability possibly by the mechanism of decreasing the threshold of excitation on the postsynaptic membrane. Strychnine is another. By its ability to interfere with the normal spinal inhibitory input to the alpha motor neurons, it produces hyperexcitability and muscular convulsions. Hexamethonium and mecamylamine can both block transmission at the synapses formed by the preganglionic and postganglionic neurons in the ganglia of the autonomic nervous system. In addition, a great number of agents are available which both stimulate or depress activity in the central nervous system. The mechanisms for their actions are largely unknown, including whether they directly stimulate or inhibit synaptic transmission or operate indirectly through metabolic changes in the neurons themselves.
In many respects the events at the neuromuscular junction (NMJ) and the neuronal synapse are similar. Both involve contacts between excitable tissues. Neuron to neuron at the neuronal synapse and neuron to skeletal muscle cell at the NMJ. Further, both presynaptic neurons release neurotransmitter at their terminal endings which diffuse across a narrow cleft to bind with receptor sites on the membrane of the postsynaptic cell, opening ion channels.
They differ in that each vertebrate skeletal muscle cell is innervated by a single neuron, whereas hundreds to thousands of neurons often converge upon a single postsynaptic neuron. Another difference lies in the fact that vertebrate neuromuscular junctions are excitatory only. There are no inhibitory junctions. Also, ACh is the only neurotransmitter to be identified with these junctions. The following descriptions refer to mammalian skeletal muscle unless otherwise indicated.
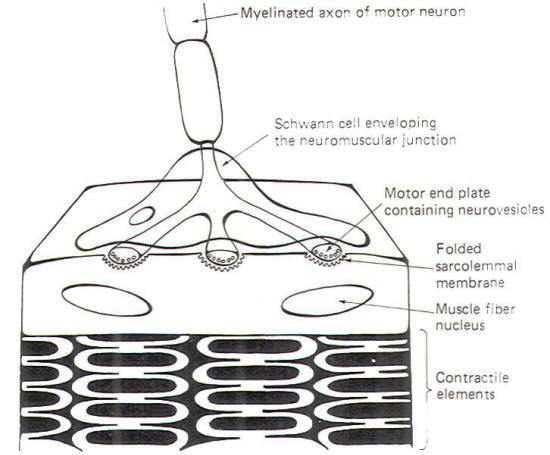 |
Fig-7
|
Skeletal muscle cells (muscle fibers) are typically innervated by largediameter myelinated neurons. These neurons have single long axons which branch into filaments numbering from a very few to several thousand. Each filament ends by forming a neuromuscular junction with a skeletal muscle cell (Fig-7). The neuronal filament terminates in a few flattened enlargements known as the motor end plates. These end plates are analogous to the presynaptic terminals in the neuronal synapse. Synaptic vesicles containing ACh are heavily concentrated in the end plates. The sarcolemmal membrane (muscle fiber membrane) beneath the end plate forms a many-folded depression. The folding of the membrane presents a greatly enlarged surface area equipped with receptor sites responsive to the ACh released by NMJ transmission
Neuromuscular junction transmission begins when an impulse reaches the motor end plate. The arrival of this impulse causes the release of ACh into the synaptic cleft, where it diffuses the short distance to the folded muscle fiber membrane. Here the ACh binds with receptor sites, causing the opening of both Na+ and K+ channels. Since the ionic distribution on either side of the sarcolemma is very similar to that which has already been observed for nerve cell membranes. Na+ will diffuse inward while K+ diffuses outward. Because the increase in Na+ permeability is greater than for K+ and because Na+ is driven by both a chemical and electrical gradient, there is a net movement of positive charges into the cell, causing it to depolarize from its normal resting state of approximately -85 mV. Once the membrane starts to depolarize, it is no longer resting and its potential is now called an end plate potential (EPP).
Action potentials are not generated on the sarcolemmal membrane directly beneath the end plates but rather on that portion of the sarcolemma adjacent to the junction. Let's take a look at how this happens. Once the EPP is established, conditions exist for the development of an electrotonic current which spreads away from the junction through the sarcoplasm (muscle fiber cytoplasm) toward the still-polarized adjacent areas of the cell. As this current spreads away from the junction, it passes out through the adjacent sarcolemma, depolarizing it to the excitation threshold and producing an action potential. This action potential then propagates as an impulse over the muscle cell, bringing about its contraction.
An important distinction between the neuronal synapse and the neuromuscular junction lies in the potency of a single synaptic discharge. A single synapse discharging once is almost never sufficient to produce an action potential in the second neuron of a neuronal synapse. We have previously described how a great many synapses firing simultaneously or a few firing repetitively at a very high rate are necessary to summate EPSPs to the excitation threshold of the postsynaptic neuron in order to produce an action potential. By contrast, a single neuromuscular junction firing once is almost always more than sufficient to produce an EPP capable of generating an action potential on the adjacent sarcolemma, bringing about impulse production and muscle fiber contraction. In fact the arrival of a single impulse at a single neuromuscular junction typically releases sufficient ACh to establish an EPP four times larger than necessary to generate the action potential. Thus we speak of a "safety factor" of 4 at the neuromuscular junction. This may seem like unnecessary waste of effort. However, if we consider that each muscle fiber receives only one neuronal input, the backup capability provided by this excess may not be out of line after all.
As is the case with the neuronal synapse, it is necessary to quickly remove the neurotransmitter after each discharge in order to keep the muscle cell from being continually stimulated, thereby eliminating the control the nerve fiber has over the contraction of the muscle cell it innervates. The great majority of ACh molecules are inactivated on the spot by the action of the enzyme acetylcholinesterase (AChE). The fraction of ACh not inactivated in this way diffuses out of the cleft or is reabsorbed by the end plate.
As with the neuronal synapse, a number of drugs are available which modify transmission at the neuromuscular junction. Curare is the classical competitive inhibitor at the junction. It competes with endogenously released ACh for the receptor sites. However. the curare-receptor site interaction does not cause depolarization and the establishment of an EPP. It thus blocks transmission of the signal from the nerve fiber to the muscle cell.
While curare is a naturally occurring drug, gallamine. benzoquinonium, and pancuronium are synthetic curarelike compounds which block neuromuscular transmission by similar mechanisms. Succinylcholine and decamethonium are also neuromuscular blocking agents but operate by a different mechanism. These compounds produce an initial depolarization of the sarcolemmal membrane which renders ACh incapable of producing a response in the already depolarized membrane. Several minutes later, as the membrane repolarizes, there is a secondary phase of decreased receptor sensitivity to ACh.
The neuromuscular blocking agents are primarily useful adjuncts to anesthesia for producing muscle relaxation. They are also useful for easing endotracheal intubation and for depressing spontaneous contraction of respiratory muscles under certain circumstances when artificial respirators are employed.
Neuromuscular transmission can be potentiated by the use of drugs which inhibit the action of the enzyme AChE. Neostigmine and physostigmine are reversible anticholinesterases. That is, they combine with AChE, for which they have a greater affinity than does ACh, and thus effectively tie up the enzyme so that it cannot degrade ACh. After a few hours, neostigmine and physostigmine uncouple from the enzyme for subsequent degradation elsewhere in the body, restoring normal function to the neuromuscular junction. Both drugs are potent anticurare agents as they allow ACh to build up in the synaptic cleft giving it a favorable competitive edge over curare for the available receptor sites. Diisopropylfluorophosphate is a potent compound which combines irreversibly with AChE, promoting long-term increases in neuromuscular transmission. It has had some therapeutic application but was primarily developed as a chemical warfare agent and is now principally of interest because of toxicological effects associated with its use as an insecticide. Its use produces a variety of signs and symptoms. including muscle fasciculations, sweating, abdominal cramps, respiratory distress, and even convulsions.
The therapeutic application of AChE inhibitors are limited by their lack of specificity since ACh levels are increased at ganglionic, postganglionic, and neuromuscular receptor sites. Neostigmine and physostigmine are primarily useful in the treatment of myasthenia gravis and glaucoma. Both are employed for the latter purpose, while neostigmine is commonly used for the treatment of myasthenia. In addition to its anticholinesterase effect, neostigmine has also been show to have a direct stimulating effect on skeletal muscle cell receptors.
The synapses made by autonomic nerve fibers with the cells of cardiac muscle, smooth muscle, and glands are more varied anatomically and chemically than are those at the neuronal synapse previously discussed and the neuromuscular junction. Nevertheless, these junctions are also characterized by the presynaptic release of neurotransmitters which diffuse to receptor sites on the effector cell membrane, producing changes in ion permeability and initiating physiological action such as muscle cell contraction or glandular activity.
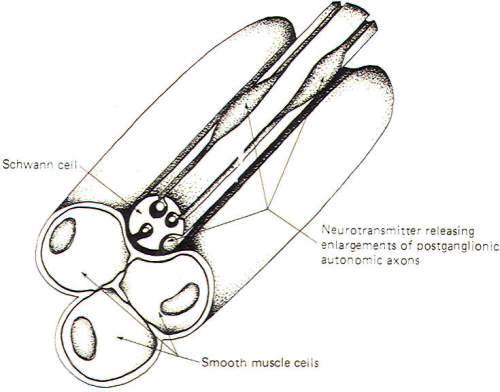 |
Fig-8
|
Fig-8 illustrates a neuroeffector junction between a nonmyelinated postganglionic autonomic fiber and smooth muscle cells. Unlike the skeletal muscle junction, here we have several points at which transmitter is released to the muscle cell membrane. The nonmyelinated axon is shown to extend out of its groove at some places in an enveloping Schwann cell and to give rise there to enlargements which contain neurotransmitter-releasing vesicles. Recognize that these axons are not myelinated by the Schwann cell but are simply enveloped in grooves formed by infoldings of the cell. Impulse transmission along the axon causes release of neurotransmitter at these points with the subsequent excitation or inhibition of the muscle cells. Like the neuronal synapse described earlier and unlike the neuromuscular junction, neuroeffector junctions can either excite or inhibit the effector cell. Postganglionic parasympathetic nerve fibers release ACh and give rise to either excitatory or inhibitory effects depending on the specific effector cell. Postganglionic sympathetic nerve fibers release either NE or ACh. The overwhelming majority of them, however, release NE. The effects of synaptically released NE are even more complex. Whether it is excitatory or inhibitory depends not only on the kind of effector cell innervated but also upon the type of receptor site located on the cell.
Acetylcholine, pilocarpine. and methacholine all directly stimulate cholinergic (ACh) receptors on autonomic effector organs. Physostigmine and neostigmine also potentiate activity at these receptors but act indirectly by their anticholinesterase activity. On the other hand, atropine is a potent antagonist at neuroeffector junctions by inhibiting the action of ACh on the receptor sites. Norepinephrine, epinephrine, isoproterenol, and phenylephrine directly stimulate adrenergic (NE) receptors. Isoproterenol is specifically a beta receptor stimulant (agonist), while phenylephrine is an alpha receptor agonist. Conversely, phentolamine and phenoxybenzamine are effective alpha antagonists that block transmission at these junctions. while propranolol is a beta blocker. For more details click here!
1 yorum:
The 6 BEST Casinos Near Carson City, CA - MapYRO
Find the best casinos in Carson 경상북도 출장마사지 City, CA. The 6 BEST 수원 출장마사지 Casinos 영주 출장샵 near Carson 동두천 출장샵 City, CA, including popular sites like 상주 출장샵 River Belle, The
Yorum Gönder